MNV Recomposition
Overview
Most annotation tools handle variants independently. The problem with this approach is that nearby variants could affect the same codon leading to a very different annotation. For example, consider the following example (Danecek, 2017):
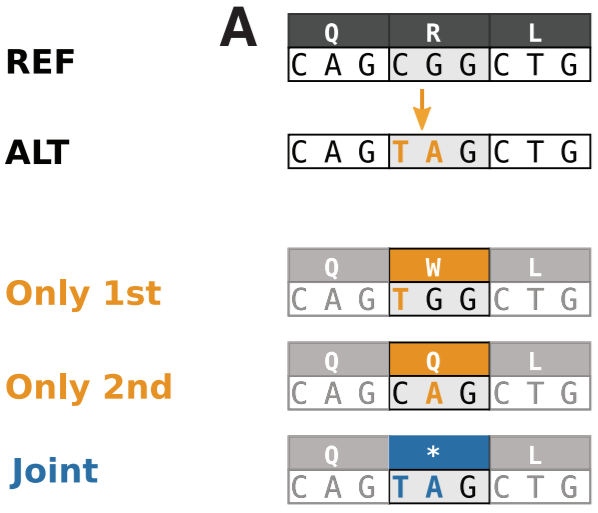
When handled independently, the two variants (C→T & G→A) would be annotated as missense annotations. However, if we consider them together, the resulting MNV would yield a stop gain.
By default, Nirvana identifies these types of cases where two or more SNVs would affect the same codon. In addition, it's able to perform this operation on VCFs containing large numbers of samples (we've tested this on 2,500+ samples using the 1000 Genomes Project VCF files).
Publication
Petr Danecek, Shane A McCarthy, BCFtools/csq: haplotype-aware variant consequences, Bioinformatics, Volume 33, Issue 13, 1 July 2017, Pages 2037–2039
Supported variant types
At the moment, Nirvana only supports recomposing multiple SNVs into an MNV. The Danecek paper makes a compelling case for supporting frameshifting variants paired with frame-restoring variants. We've also received requests for supporting the recomposition of an SNV with insertions and deletions. While this is something we've looked into, it represents functionality that many of our clinical customers are not yet comfortable with.
Criteria
Nirvana will recompose a set of SNVs if two or more SNVs are located in the same codon for any codon in any of the overlapping transcripts.
The following criteria must also be met for at least one sample:
- Genotypes are provided for the VCF variants and all variants are in phase or homozygous variant.
- All the available phase set IDs are the same (homozygous variants are available to all phase sets)
- The genotype ploidy for all the variants are the same.
- No unsupported variant type (i.e. insertion or deletion) overlaps the recomposed variants
- The first and last base in at least one of the recomposed alleles must be non-reference.
Examples
During variant recomposition, if two SNVs affect the same codon, it becomes the seed codon. If there are SNVs in the adjacent codons, they will be aggregated into the seed codon.
Three SNVs in two adjacent codons. The recomposed alternate allele is
ATAG: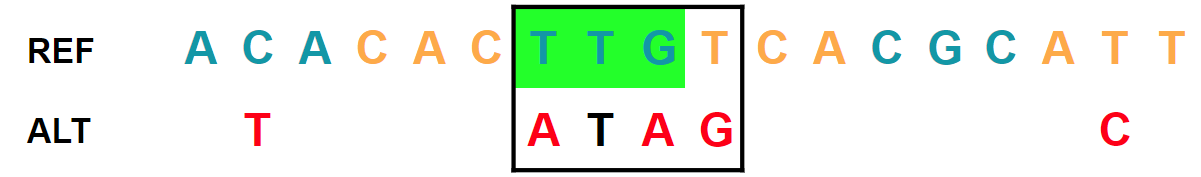
Three SNVs in two adjacent codons (larger distance). The recomposed alternate allele is
ATATCC: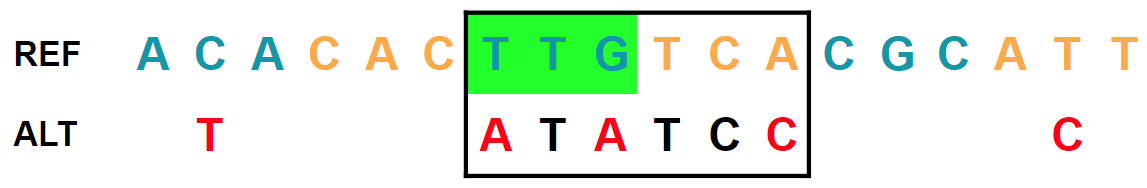
Nirvana can use multiple reading frames to aggregate the seed codon. In this example, the seed codon is highlighted in green. If we look at reading frame 1, we see that the T→A variant occurs in the
ACTcodon. The adjacent codon to the left also has a variant C→T. As a result, there can be up to four bases between SNVs when aggregating the flanking codons. The recomposed alternate allele isTTCACATAGCACTCAC: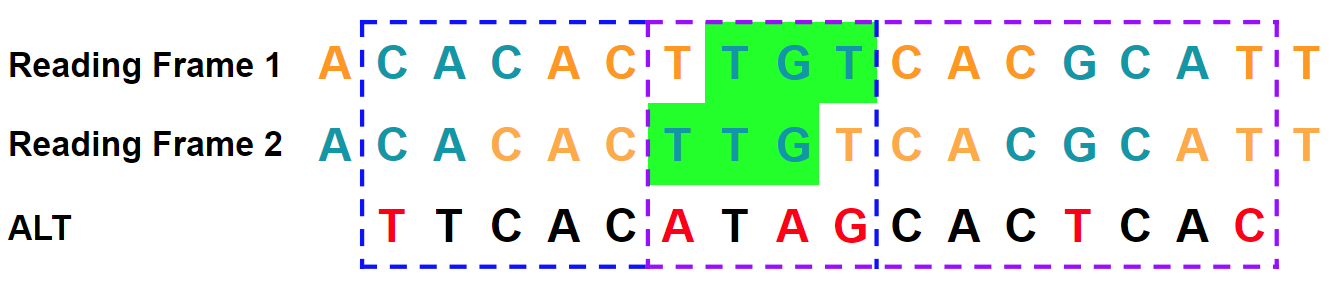
Nothing will be recomposed if there's no seed codon:

Multiple Samples
Recomposing variants while handling multiple samples can be complex. The recomposition criteria described above often leads to sample-specific recomposed variants. Here we show the recomposition of three variants with sample-specific criteria marked in bold:
| POS | REF | ALT | Sample 1 | Sample 2 | Sample 3 | |
|---|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | C | 0|1 | 0|1 | 1|1 |
| Decomposed Variant 2 | 101 | C | G | 0/1 | 1|1 | 0|0 |
| Decomposed Variant 3 | 102 | T | A | 1|1 | . | 0|1 |
| Recomposed Variant 1 | 100 | AC | AG, CG | . | 1|2 | . |
| Recomposed Variant 2 | 100 | ACT | CCT, CCA | . | . | 1|2 |
In the example above, the heterozygous genotype in sample 1 at position 101 would prevent the MNVs from being recomposed. Similarly, the unknown genotype for sample 2 at position 102 would produce a smaller MNV than the one expressed for sample 3.
Phase Sets
Homozygous variants, same phase set
Recomposed phase set becomes . since homozygous variants belong to all phase sets.
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | T | 1|1 | 567 |
| Decomposed Variant 2 | 101 | C | G | 1|1 | 567 |
| Recomposed Variant | 100 | AC | TG | 1|1 | . |
Mixing phased and unphased variants
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | T | 0|1 | 567 |
| Decomposed Variant 2 | 101 | C | G | 1/1 | . |
| Recomposed Variant | 100 | AC | AG,TG | 1|2 | 567 |
Variants in different phase sets
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | T | 0|1 | 567 |
| Decomposed Variant 2 | 101 | C | G | 1|1 | 890 |
| Recomposed Variant | 100 | AC | AG,TG | 1|2 | . |
Unphased homozygous variants
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | T | 1/1 | . |
| Decomposed Variant 2 | 101 | C | G | 1/1 | . |
| Recomposed Variant | 100 | AC | TG | 1/1 | . |
Homozygous variants are not commutative
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Decomposed Variant 1 | 100 | A | T | 0|1 | 567 |
| Decomposed Variant 2 | 101 | C | G | 1|1 | 567 |
| Decomposed Variant 3 | 102 | G | T | 0|1 | 890 |
In this example, the homozygous variant at position 101 cannot bridge the gap between other two variants since there could be a switching error between phase sets 567 & 890. As a result, we have to create two overlapping MNVs:
| POS | REF | ALT | Genotype | Phase Set | |
|---|---|---|---|---|---|
| Recomposed Variant 1 | 100 | AC | AG, TG | 1|2 | 567 |
| Recomposed Variant 2 | 101 | CG | GG, GT | 1|2 | 890 |
Conflicting Genotypes
JSON Output
Given the following VCF entries:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT S1 S2 S3
chr1 12861477 . T C . PASS . GT:PS 0/0:. 0/0:. 0|1:12861477
chr1 12861478 . G A . PASS . GT:PS 0/0:. 0/0:. 0|1:12861477
Each original variant would be annotated as usual. The difference is that both will now have a isDecomposedVariant flag set to true in addition to an entry in the linkedVids field that points to the new MNV:
{
"chromosome":"chr1",
"position":12861477,
"refAllele":"T",
"altAlleles":[
"C"
],
"filters":[
"PASS"
],
"samples":[
{
"genotype":"0/0",
},
{
"genotype":"0/0",
},
{
"genotype":"0|1",
}
],
"variants":[
{
"vid":"1-12861477-T-C",
"chromosome":"chr1",
"begin":12861477,
"end":12861477,
"refAllele":"T",
"altAllele":"C",
"variantType":"SNV",
"isDecomposedVariant":true,
"linkedVids":[
"1-12861477-TG-CA"
],
"hgvsg":"NC_000001.11:g.12861477T>C",
"transcripts":[ ... ]
}
]
},
{
"chromosome":"chr1",
"position":12861478,
"refAllele":"G",
"altAlleles":[
"A"
],
"filters":[
"PASS"
],
"samples":[
{
"genotype":"0/0",
},
{
"genotype":"0/0",
},
{
"genotype":"0|1",
}
],
"variants":[
{
"vid":"1-12861478-G-A",
"chromosome":"chr1",
"begin":12861478,
"end":12861478,
"refAllele":"G",
"altAllele":"A",
"variantType":"SNV",
"isDecomposedVariant":true,
"linkedVids":[
"1-12861477-TG-CA"
],
"hgvsg":"NC_000001.11:g.12861478G>A",
"transcripts":[ ... ]
}
]
}
The recomposed variant gets a separate entry where the isRecomposedVariant flag is set to true and the linkedVids field links to the constituent SNVs:
{
"chromosome":"chr1",
"position":12861478,
"refAllele":"G",
"altAlleles":[
"A"
],
"filters":[
"PASS"
],
"samples":[
{
"genotype":"0/0",
},
{
"genotype":"0/0",
},
{
"genotype":"0|1",
}
],
"variants":[
{
"vid":"1-12861478-G-A",
"chromosome":"chr1",
"begin":12861478,
"end":12861478,
"refAllele":"G",
"altAllele":"A",
"variantType":"SNV",
"isDecomposedVariant":true,
"linkedVids":[
"1-12861477-TG-CA"
],
"hgvsg":"NC_000001.11:g.12861478G>A",
"transcripts":[ ... ]
}
]
}
Recomposed QUAL, FILTER, and GQ
Although the example above does not demonstrate it, Nirvana tries to set the quality score, filter, and genotype quality (GQ) for the recomposed variant. The QUAL score is calculated to be the minimum QUAL score for all the constituent SNVs. The same method is used for the genotype quality (GQ) scores. For the filters field, PASS will be used if all constituent variants passed their filters, otherwise we set it to FilteredVariantsRecomposed.